e+
e−
2D > 3D
C
H
N
O
P
S
F
Cl
Br
I
...
| Formula | |
| Masa moleculara | |
| Hydrogen - donatori | |
| Hydrogen - acceptori |
| Formula | |
| Masa moleculara | |
| Hydrogen - donatori | |
| Hydrogen - acceptori |

MolView consists of two main parts, a structural formula editor and a 3D model viewer. The structural formula editor is surround by three toolbars which contain the tools you can use in the editor. Once you’ve drawn a molecule, you can click the 2D to 3D button to convert the molecule into a 3D model which is then displayed in the viewer. Below is a list of all sketch tools.


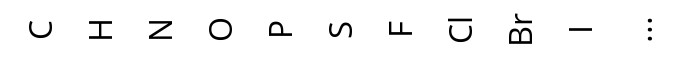
In this toolbar you can select from a number of elements, you can also pick an element from the periodic table using the last button. You can use the element to create new atoms or modify existing atoms.

You can load molecules from large databases like PubChem and RCSB using the search form located on the left side of the menu-bar. Just type what you are looking for and a list of available molecules will appear.
You can also click on the dropdown button next to the search field to select a specific database. This will perform a more extensive search on the selected database. Currently, three big databases are supported:
The Tools menu contains several utility functions which are listed below.
You can embed or share a specific compound, macromolecule or crystal using the provided URL or HTML code. Note that the linked structure is the one which is currently displayed in the model window. You can also copy the URL from the address bar in order to link to the current structure.
Export options:
This collects and displays information about the structural formula.
This shows a new layer where you can view molecular spectra of the current structural formula (loaded from the Sketcher) More details are covered in the Spectroscopy chapter.
This redirects you to the web-page for the current 3D model on the website of its source database (except when the model is resolved using the Chemical Identifier Resolver)
These functions allow you to perform some advanced searches through the PubChem database using the structural formula from the sketcher.
You can open the Spectroscopy view via Tools > Spectroscopy. You can view three kinds of molecular spectra.
The Model menu contains some general functions for the 3D model.
This function sets the model position, zoom and rotation back to default.
You can choose from a list of different molecule representations including; ball and stick, stick, van der Waals spheres, wireframe and lines. Macromolecules are automatically drawn using ribbons.
You can switch between a black, gray or white background. The default background is black (exported images from GLmol or ChemDoodle have a transparent background)
You can choose from three different render engines: GLmol, Jmol and ChemDoodle. GLmol is used as default render engine. GLmol and ChemDoodle are based on WebGL, a browser technology to support 3D graphics. If WebGL is not available in your browser, Jmol will be used for all rendering.
MolView automatically switches to:
You might want to switch back to GLmol when you do no longer need Jmol or ChemDoodle since GLmol has a better performance.
Note that macromolecules are drawn slightly different in each engine. ChemDoodle provides the finest display. You should, however, avoid using ChemDoodle for very large macromolecules.
You can rotate, pan and zoom the 3D model. Use the right button for rotation, the middle button for translation (except for ChemDoodle) and the scrollwheel for zooming. On touch devices, you can rotate the model with one finger and scale the model using two fingers.
You can load an array of crystal cells (2x2x2 or 1x3x3) or a single unit cell when viewing crystal structures.
When you are viewing large structures, like proteins, it can be useful to hide a certain part using fog or a clipping plane. GLmol offers a few options to do this.
The Jmol menu offers some awesome Jmol-only functions and calculations.
Clears all executed calculations and measurements.
Enables High Quality rendering in Jmol (enabled by default on fast devices) When turned off, anti-aliasing is disabled and the model is drawn using lines while transforming it.
You can perform the following Jmol calculations in Jmol:
You can measure distance, angle and torsion using Jmol. You can activate and deactivate one of these measurement types via the Jmol menu.
Note that in some cases, the resolved 3D model is only an approach of the real molecule, this means you have to execute an Energy minimization in order to do reliable measurements.
Poti utiliza adresa URL pentru a te conecta la modelul 3D actual.